comp_chem_utils¶
This directory contains the most important and fundamental modules of the comp_chem_py suite.
utils¶
Collection of simple functions useful in computational chemistry scripting.
Many of the following functions are used to make operations on xyz coordinates
of molecular structure. When refering to xyz_data bellow, the following
structures (also used in molecule_data) is assumed:
atom 1 label and corresponding xyz coordinate
atom 2 label and corresponding xyz coordinate
: : :
atom N label and corresponding xyz coordinate
For example the xyz_data of a Hydrogen molecule along the z-axis
should be passed as:
>>> xyz_data
[['H', 0.0, 0.0, 0.0], ['H', 0.0, 0.0, 1.0]]
-
vel_auto_corr(vel, max_corr_index, tstep)[source]¶ Calculate velocity autocorrelation function.
Parameters: - vel (list) – The velocities along the trajectory are given as
a list of
np.array()of dimensions N_atoms . 3. - max_corr_index (int) – Maximum number of steps to consider for the auto correlation function. In other words, it corresponds to the number of points in the output function.
- vel (list) – The velocities along the trajectory are given as
a list of
-
get_lmax_from_atomic_charge(charge)[source]¶ Return the maximum angular momentum based on the input atomic charge.
This function is designed to return LMAX in a CPMD input file.
Parameters: charge (int) – Atomic charge. Returns: ‘S’ for H or He; ‘P’ for second row elements; and ‘D’ for heavier elements.
-
get_file_as_list(filename, raw=False)[source]¶ Read a file and return it as a list of lines (str).
By default comments (i.e. lines starting with #) and empty lines are ommited. This can be changed by setting
raw=TrueParameters: - filename (str) – Name of the file to read.
- raw (bool, optional) – To return the file as it is,
i.e. with comments and blank lines. Default
is
raw=False.
Returns: A list of lines (str).
-
make_new_dir(dirn)[source]¶ Make new empty directory.
If the directory already exists it is erased and replaced.
Parameters: dirn (str) – Name for the new directory (can include path).
-
center_of_mass(xyz_data)[source]¶ Calculate center of mass of a molecular structure based on xyz coordinates.
Parameters: xyz_data (list) – xyz atomic coordinates arranged as described above. Returns: 3-dimensional np.array()containing the xyz coordinates of the center of mass of the molecular structure. The unit of the center of mass matches the xyz input units (usually Angstroms).
-
change_vector_norm(fix, mob, R)[source]¶ Scale a 3-D vector defined by two points in space to have a new norm R.
The input vector is defined by a fix position in 3-D space
fix, and a mobile positionmob. The function returns a new mobile position such that the new vector has the norm R.Parameters: - fix (np.array) – xyz coordinates of the fix point.
- mob (np.array) – Original xyz coordinates of the mobile point.
- R (float) – Desired norm for the new vector.
Returns: The new mobile position as an
np.array()of dimenssion 3.
-
change_vector_norm_sym(pos1, pos2, R)[source]¶ Symmetric version of change_vector_norm function.
In other word both positions are modified symmetrically.
-
get_rmsd(xyz_data1, xyz_data2)[source]¶ Calculate RMSD between two sets of coordinates.
The Root-mean-square deviation of atomic positions is calculated as
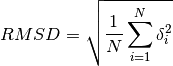
Where
\delta_iis the distance between atom i inxyz_data1and inxyz_data2.Parameters: - xyz_data1 (list) – List of atomic coordinates for the first structure arranged as described above for xyz_data.
- xyz_data2 (list) – Like
xyz_data1but for the second structure.
Returns: The RMSD (float).
-
get_distance(xyz_data, atoms, box_size=None)[source]¶ Calculate distance between two atoms in xyz_data.
Parameters: - xyz_data (list) – xyz atomic coordinates arranged as described above.
- atoms (list) – list of two indices matching the two rows of the xyz_data for wich the distance should be calculated.
Returns: Distance between the two atoms in the list as a
float, the unit will match the input unit in thexyz_data.
-
get_angle(xyz_data, atoms)[source]¶ Calculate angle between three atoms in xyz_data.
Parameters: - xyz_data (list) – xyz atomic coordinates arranged as described above.
- atoms (list) – list of three indices matching the rows of the xyz_data for wich the angle should be calculated.
Returns: Angle between the three atoms in the list as a
floatin degrees.
-
get_dihedral_angle(table, atoms)[source]¶ Calculate dihedral angle defined by 4 atoms in xyz_data.
It relies on the praxeolitic formula (1 sqrt, 1 cross product).
Parameters: - xyz_data (list) – xyz atomic coordinates arranged as described above.
- atoms (list) – list of 4 indices matching the rows of the xyz_data for wich the dihedral angle should be calculated.
Returns: Dihedral angle defined by the 4 atoms in the list as a
floatin degrees.
conversions¶
Functions and constants to convert quantities from and to different units.
When executed directly, calls the two test functions:
print_constants()test_conversion()
-
EV_TO_JOULES= 1.602176565e-19¶ Energy conversion from eV to J.
By definition an electron-volt is the amount of energy gained (or lost) by the charge of a single electron moving across an electric potential difference of one volt. So 1 eV is 1 volt (1 joule per coulomb, 1 J/C) multiplied by the elementary charge.
-
AU_TO_JOULES= 4.359744340736074e-18¶ Energy conversion from Hartree (a.u.) to J.
1 Hartree (a.u.) is defined as
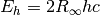
-
CAL_TO_JOULES= 4.184¶ Energy conversion from cal to J.
1 calorie is defined as the amount of heat energy needed to raise the temperature of one gram of water by one degree Celsius at a pressure of one atmosphere. The thermochemical calorie (used here) is defined to be exactly 4.184 J.
-
KCALMOL_TO_JOULES= 6.947694845598683e-21¶ Energy conversion from kcal.mol-1 to J.
From the definition of the calorie.
-
AU_TO_KELVIN= 315775.04291721934¶ Conversions from a.u. to Kelvin when calculating kinetic temperature.
For a given kinetic energy in a.u. E_kin, we can obtain the kinetic temperature in Kelvin as:
Temp = 2.0 * E_kin * AU_TO_KELVIN / NDOF,
Where NDOF is the Number of Degrees Of Freedom.
-
AU_TO_S= 2.4188843280245007e-17¶ Time conversion from a.u. to seconds.
-
AU_TO_FS= 0.024188843280245006¶ Time conversion from a.u. to femto-seconds.
-
AU_TO_PS= 2.4188843280245007e-05¶ Time conversion from a.u. to pico-seconds.
-
BOHR_TO_ANG= 0.52917721092¶ Distance conversion from Bohr (a.u.) to Angstroms.
-
ATOMIC_MASS_AU= 1822.8884847700401¶ Atomic mass unit expressed in atomic units.
-
SPEC_TO_SIGMA= 1667562945278699.8¶ Conversion constant between a spectral function
Sexpressed in seconds (reciprocal angular frequency unit) and the absorption cross section\sigmaexpressed inAngstroms^2.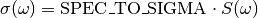
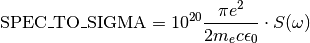
For more details see the documentation of the
spectrummodule.
-
SIGMA_TO_EPS= 26153.8273148873¶ Conversion constant between an absorption cross section
\sigmaexpressed inAngstroms^2and the extinction coefficient expressed inM^{-1} . cm^{-1}.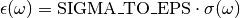
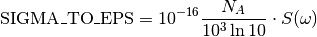
For more details see the documentation of the
spectrummodule.
-
convert(X, from_u, to_u)[source]¶ Convert the energy value
Xfrom the unitfrom_uto the unitto_u.Parameters: - X (float) – Energy value to convert.
- from_u (str) – Unit of the input energy value
Xgiven as one of the keys of the dictionaryconvert_to_joules. - to_u (str) – Unit of the output energy value
Xgiven as one of the keys of the dictionaryconvert_to_joules.
Returns: The
Xvalue is returned expressed in theto_uunit.Example
>>> from comp_chem_utils import conversions as c >>> c.convert(1.0, 'WAVE LENGTH: nm', 'ENERGY: eV') 1239.8419292004205
physcon¶
Library of physical constants.
This is coming from an old version of https://github.com/georglind/physcon.git
>>> import comp_chem_utils.physcon as pc
>>> pc.help()
Available functions:
[note: key must be a string, within quotes!]
value(key) returns value (float)
sd(key) returns standard deviation (float)
relsd(key) returns relative standard deviation (float)
descr(key) prints description with units
:
Available global variables:
alpha, a_0, c, e, eps_0, F, G, g_e, g_p, gamma_p, h, hbar, k_B
m_d, m_e, m_n, m_p, mu_B, mu_e, mu_N, mu_p, mu_0, N_A, R, sigma, u
:
Available keys:
['alpha', 'alpha-1', 'amu', 'avogadro', 'bohrmagn', 'bohrradius', 'boltzmann', 'charge-e', 'conduct-qu', 'dirac', 'elec-const', 'faraday', 'gas', 'gfactor-e', 'gfactor-p', 'gravit', 'gyromagratio-p', 'josephson', 'lightvel', 'magflux-qu', 'magn-const', 'magnmom-e', 'magnmom-p', 'magres-p', 'mass-d', 'mass-d/u', 'mass-e', 'mass-e/u', 'mass-mu', 'mass-mu/u', 'mass-n', 'mass-n/u', 'mass-p', 'mass-p/u', 'nuclmagn', 'planck', 'ratio-me/mp', 'ratio-mp/me', 'rydberg', 'stefan-boltzm']
-
value(key)[source]¶ Returns the value (float) of the physical constant corresponding to the input key.
Parameters: key (str) – single word description of an available constant. See help()to see available keys.
-
sd(key)[source]¶ Returns the standard deviation (float) of the physical constant corresponding to the input key.
spectrum¶
Collection of functions to read, calculate, and plot electronic spectra.
-
class
Search(fname, search_str, col_id, offset=0, trim=(0, None), cfac=1.0)[source]¶ Class for defining how to search spectrum information in an computational chemistry output file.
The information needed are excitation energies and oscillator strength. The assumption here is that in the output file this information can be located from a string
search_str.The data to be read can be shifted from the
search_strby anoffsetnumber of lines. It is then found in a specificcol_idinside the line which is splitted based on blank characters. It can be trimmed bytrimand converted bycfac.Example
Possible output file (named
'my_output'):Excited State E=1.0000cm-1 f=0.00000
In that case to extract the energy in eV we would have to do:
>>> from comp_chem_utils.spectrum import Search >>> my_search_obj = Search('my_output', 'Excited State', 0, offset=1, trim=(2,7), cfac=1.2398e-4) >>> my_search_obj.get_all()[0] 0.00012398
Parameters: - fname (str) – Name of the output file containing the spectrum information. Can include path to file.
- search_str (str) – String that will be search in the output file to locate the data to be extracted.
- col_id (int) – The line matching the search string is splitted into a list with
str.split()and the elementcol_idof the list is extracted. - offset (int, optional) – In case the line of interest is not the line with
search_strtheoffsetcan be used to shift to a line above (negative offset) or bellow the matching line. Default value is 0. - trim (tuple) – In case the element
col_idextracted from the line contains more than the desired data. A subset of the string can be extracted with trim. See example. Default is(0,None), i.e. it takes the whole element. - cfac (float, optional) – In case the extracted data is not in the desired unit, it can be
multiplied by
cfacto convert it. Default value is 1.0.
-
search_exc(kind, output)[source]¶ Get excitation energies from an output file.
The
kindof output file is used to determine the parameters to be used to set theSearchclass.
-
search_osc(kind, output)[source]¶ Get oscillator strengts from an output file.
The
kindof output file is used to determine the parameters to be used to set theSearchclass.
-
print_spectrum(exc_ener, strength, output)[source]¶ Print table summary of Excitation energies and oscillator strengths.
-
read_spectrum(output, kind, verbose=False)[source]¶ Read an output file and parse it to find excitation energies and oscillator strengths.
Note
If the spectrum data is written as a table, i.e. in the following format:
THIS IS A TABLE OF SPECTRUM DATA: E (eV) f 1.000 0.000 1.000 0.000 : :
Then the
read_table_spectrum()function should be used instead.Parameters: - output (str) – Name of the output file containing the spectrum information. Can include path to file.
- kind (str) – Kind of output, e.g. ‘gaussian’ or ‘lsdalton’. This is used in the
search_exc()andsearch_osc()to set the parameters of theSearchclass. Newkindhave to be implemented in those functions. - verbose (bool, optional) – If
Truethe extracted data will be printed out. Default isFalse.
Returns: exc, osc
Excitation energies and oscillator strengths are returned in the form of the two 1-D
np.array(),excandosc.
-
read_table_spectrum(output, search_str='', offset=0, pos_e=0, pos_f=1, verbose=False)[source]¶ Read spectrum data arranged as a table in the output file.
Parameters: - output (str) – Name of the output file containing the spectrum information. Can include path to file.
- search_str (str, optional) – String that will be search in the output file to locate the data to be extracted. If it’s not provided, the output is assumed to contain only raw data.
- offset (int, optional) – Number of lines to skip after the matching line before the table starts. Default is 0, which means that the table is assumed to start right after the matching line.
- pos_e (int, optional) – Column index for the excitation energies. Default is 0.
- pos_f (int, optional) – Column index for the oscillator strengths. Default is 1.
- verbose (bool, optional) – If
Truethe extracted data will be printed out. Default isFalse.
Returns: exc, osc
Excitation energies and oscillator strengths are returned in the form of the two 1-D
np.array(),excandosc.
-
spectral_function(exc, osc, unit_in='ENERGY: eV', nconf=1, fwhm=None, ctype='lorentzian', x_range=None, x_reso=None, raw=False)[source]¶ Calculate the spectral function from theoretical data (excitation energies and oscillator strengths).
Note
It is not recommended to use this function directly. Instead the
plot_spectrum()function should be used which serves as a wrapper and gives more flexibility on the output data.The spectral function is calculated as
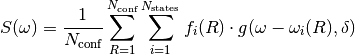
This expression is very well descrined in, e.g. [Barbatti2010a]. (f_i, omega_i) is the pair of input excitation energies and oscillator strengths, while omega is the incident frequency. The spectral line shape function is g which depends on the Full Width at Half Maximum delta, it is expressed in reciprocal angular frequency units, i.e. seconds (per molecules or structure).
Parameters: - exc – Input excitation energies given in a one dimenssional
np.array(). - osc – Input oscillator strengths given in a one dimenssional
np.array(). - unit_in (str, optional) – String describing the unit used for the input
excitation energies. The string must correspond to one of the keys of
the dictionary
convert_to_joulesin theconversionsmodule. Default is'ENERGY: eV'. - nconf (int, optional) – Total number of conformations used in the input data. This is used for normalization to a single structure. Default is 1.
- fwhm (float, optional) – Full width at Half Maximum used in the convolution function.
It must be given in the same units as
unit_in. The Default value isNone, which will be latter changed to correspond to 0.1 eV. - ctype (str, optional) – Defines the type of convolution. Either
'lorentzian'which is default or'gaussian'. - x_range (list, optional) – This is a two-value list defining the range of energy data for which
the spectral function has to be calculated. It should be given in the same units
as
unit_in. It is default toNonewhich will be latter changed to appropriate values related to the FWHM. - x_reso (int, optional) – Resolution of the spectral function given as the number of grid points
per energy unit (
unit_in). The default value isNone, which will be latter changed to correspond to 100 pts per eV. - raw (bool, optional) – When
True, the input excitation energies will not be converted to reciprocal angular frequency units. The output spectral function will thus be in reciprocalunit_inunits and the user has to be careful for what is done to the output afterwards. The default isFalse.
Returns: xpts, ypts
Those are two
np.array()containing the grid points required to plot the spectral function. No matter the unit of the input energies, (unit_in). The spectral function (in ypts) is expressed in reciprocal angular frequency (seconds). While the xpts values are expressed inunit_inenergy unit.- exc – Input excitation energies given in a one dimenssional
-
temperature_effect(E, unit_in='ENERGY: eV', temp=None)[source]¶ Calculate tenperature factor for the absorption cross section.
The temperature effect is calculated as
![1.0 - \exp [ - E / (k_{B} T) ]](_images/math/70831a9e402162fbdeb33e40357ca3d187d3fa8d.png)
where E is the input energy which will be converted to Joules.
Parameters: - E (float) – Input energy or frequency expressed in
unit_in. - unit_in (str, optional) – String describing the unit used for the input
excitation energies. The string must correspond to one of the keys of
the dictionary
convert_to_joulesin theconversionsmodule. Default is'ENERGY: eV'. - temp (float, optional) – Working temperature in Kelvin. The default value is
None, which means that no temperature effect will be calculated.
Returns: The temperature effect as a float. If
temp=None, the value returned is 1.0.- E (float) – Input energy or frequency expressed in
-
cross_section(xpts, ypts, unit_in='ENERGY: eV', temp=None, refraction=1.0)[source]¶ Calculate the absorption cross-section from the spectral function.
The cross section is just a scaled version of the spectral function that might depend on temperature and the refraction index of the medium.
Note
It is not recommended to use this function directly. Instead the
plot_spectrum()function should be used which serves as a wrapper and gives more flexibility on the output data.The
xpts, andyptsinput data are expected to come directly from a call tospectral_function().Parameters: - xpts (np.array) – Grid points corresponding to the x-axis of the spectral
function. They are expressed in
unit_in. - ypts (np.array) – Grid points corresponding to the y-axis of the spectral function. They have to be given in reciprocal angular frequency units (seconds).
- unit_in (str, optional) – String describing the unit used for the input
excitation energies. The string must correspond to one of the keys of
the dictionary
convert_to_joulesin theconversionsmodule. Default is'ENERGY: eV'. - temp (float, optional) – Working temperature in Kelvin. The default value is
None, which means that no temperature effect will be calculated. - refraction (float, optional) – The refraction index of the medium. Default value is 1.0.
Returns: The absorption cross section is returned in
Angstrom^2 . molecule^{-1}. It is calculated from the spectral function as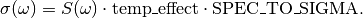
where the temp_effect comes from
temperature_effect(),SPEC_TO_SIGMAis the main conversion constant fromconversions, and n is the refraction index.The absorption cross section is returned as grid points in an
np.array().- xpts (np.array) – Grid points corresponding to the x-axis of the spectral
function. They are expressed in
-
plot_stick_spectrum(exc, osc, color=None, label=None)[source]¶ Plot a stick spectrum from theoretical data.
The raw excitation energies and oscillator strength are used to make a stick spectrum.
The color and label arguments should be specified in order to ensure a uniform color for all the sticks.
Parameters: - exc – Input excitation energies given in a one dimenssional
np.array(). - osc – Input oscillator strengths given in a one dimenssional
np.array(). - color (optional) – color code used in matplotlib.
- label (optional) – label to describe the data.
Returns: Handle object comming out of the plt call that can be used for the legend.
- exc – Input excitation energies given in a one dimenssional
-
plot_spectrum(exc, osc, unit_in='ENERGY: eV', nconf=1, fwhm=0.1, temp=0.0, refraction=1.0, ctype='lorentzian', x_range=None, x_reso=None, kind='CROSS_SECTION', with_sticks=False, plot=True)[source]¶ Plot a spectrum based on theoretical data points.
This is the main function of the module that should be used to calculate and plot electronic spectra.
Parameters: - exc – Input excitation energies given in a one dimenssional
np.array(). - osc – Input oscillator strengths given in a one dimenssional
np.array(). - unit_in (str, optional) – String describing the unit used for the input
excitation energies. The string must correspond to one of the keys of
the dictionary
convert_to_joulesin theconversionsmodule. Default is'ENERGY: eV'. - nconf (int, optional) – Total number of conformations used in the input data. This is used for normalization to a single structure. Default is 1.
- fwhm (float, optional) – Full width at Half Maximum used in the convolution function.
It must be given in the same units as
unit_in. The Default value isNone, which will be latter changed to correspond to 0.1 eV. - temp (float, optional) – Working temperature in Kelvin. The default value is
None, which means that no temperature effect will be calculated. - refraction (float, optional) – The refraction index of the medium. Default value is 1.0.
- ctype (str, optional) – Defines the type of convolution. Either
'lorentzian'which is default or'gaussian'. - x_range (list, optional) – This is a two-value list defining the range of energy data for which
the spectral function has to be calculated. It should be given in the same units
as
unit_in. It is default toNonewhich will be latter changed to appropriate values related to the FWHM. - x_reso (int, optional) – Resolution of the spectral function given as the number of grid points
per energy unit (
unit_in). The default value isNone, which will be latter changed to correspond to 100 pts per eV. - kind (str, optional) –
String describing the type of spectrum that should be calculated. It has to be one of the following:
'STICKS': 'Oscillator strength [Arbitrary units]', 'SPECTRAL_FUNC': 'Spectral function [s. $\cdot $ molecules$^{-1}$]', 'CROSS_SECTION': 'Cross section [$\AA^2 \cdot $ molecules$^{-1}$]', 'EXPERIMENTAL': 'Molar absorptivity [M$^{-1} \cdot $cm$^{-1}$]'
The default is
kind='CROSS_SECTION'. - with_sticks (bool, optional) – If
True, a stick spectrum will be plotted on top of what is required by thekindkeyword. (Default isFalse). Will affect only whenplot==True. - plot (bool, optional) – To control wether to plot or not the spectrum. The default is to plot it.
Returns: xpts, ypts
Those are two
np.array()containing the grid points required to plot the desired spectrum. The xpts array contains the x-axis values inunit_inunit, while the ypts array contains the spectrum intensity with units depending on the chosenkind.By default or if
plot=True, the function will plot the desired spectrum and show it using thematplotlibtool.- exc – Input excitation energies given in a one dimenssional
cpmd_utils¶
Collection of functions to read and manipulate CPMD output files
-
read_standard_file(fn)[source]¶ Read standard trajectory file and export it.
Here a trajectory file is understood as a file arranged as columns in which the first column contains the step indices and the others any type of informations.
For example the ENERGIES or SH_STATE.dat files enter in this category.
Parameters: fn (str) – Name of the trajectory file. Returns: steps, info steps is as list of step indices (int) as read from the first column of the trajectory file, while info is an array (
np.array()) of floats containing the rest of the information.
-
read_TRAJEC_xyz(fn)[source]¶ Read TRAJEC.xyz file and export it.
An xyz type information is read for each step in the trajectory file, where xyz info is assumed to have the following format:
number of atoms title line = step index atom 1 label and corresponding xyz coordinate atom 2 label and corresponding xyz coordinate : : : atom N label and corresponding xyz coordinate
Parameters: fn (str) – Name of the TRAJEC.xyz file. Returns: steps, traj_xyz - steps (list): List of step indices (int) as read from
- the title line of each xyz_data block.
- traj_xyz (list): List of xyz_data blocks. Each block
- is itself a list of the lines containing the coordinates in the xyz format. Each line is a 4 item list with first index the atom symbol and the last 3 items are the xyz coodinates.
-
read_GEOMETRY_xyz(fn)[source]¶ Like read_TRAJEC_xyz for a single geometry and without the step index.
-
write_TRAJEC_xyz(steps, traj_xyz, output)[source]¶ Write a TRAJEC.xyz file (CPMD style) to the output file.
Parameters: - steps (list) – Step indices. See read_TRAJEC_xyz() function.
- traj_xyz (list) – xyz data. See read_TRAJEC_xyz() function.
- output (str) – Name (and path) fo the file in which the information will be written.
-
split_TRAJEC_data(steps, traj_xyz, verbose=True, interactif=True, write_xyz=False, start_i=1, nstep=100, delta=None, dt=5, name='')[source]¶ Select equidistant xyz data snapshot from trajectory data.
From a starting index, a number of snapshots and a number of steps between each snapshot. A new trajectory data is generated with only a subset of steps.
Parameters: - steps (list) – Step indices. See read_TRAJEC_xyz() function.
- traj_xyz (list) – xyz data. See read_TRAJEC_xyz() function.
- verbose (bool, optional) – Print more information while running.
Default
is True. - interactif (bool, optional) – Let the user chose the parameters
interactively. Default
is True. - write_xyz (bool, optional) – Write an xyz file to disk for every step.
Default
is False. - start_i (int, optional) – Step index for the first snapshot. Default is 1
- nstep (int, optional) – Total number of final snapshots. Default is 100.
- delta (int, optional) – Number of steps between two snapshots.
Default is
None. It will be changed to the maximum possible value depending on other paramters. - dt (int, optional) – Time step used in MD [in a.u.] (to provide print out the actual time between the snapshots). Default is 5 a.u.
- name (str, optional) – String/Title to be used in xyz filename
in case
write_xyz = True.
Returns: new_steps, new_traj_xyz
Just a subset of
stepsandtraj_xyz.
-
read_FTRAJECTORY(fn, forces=True, verbose=False)[source]¶ Read the FTRAJECTORY file from a CPMD run and export it.
Three different formats can be read.
The TRAJECTORY file (with
forces=False):Column 0: step index Column 1-3: xyz coordinates Column 4-6: velocities
The FTRAJECTORY file (with
forces=True):Column 0: step index Column 1-3: xyz coordinates Column 4-6: velocities Column 7-9: forces
The FTRAJECTORYMTS file (with
forces=True, this file is not produced anymore):Column 0: step index Column 1-3: low level forces Column 4-6: high level forces Column 7-9: xyz coordinates
-
read_ENERGIES(fn, code, factor=1, HIGH=True)[source]¶ Read ENERGIES file from CPMD and return data based on code:
Parameters: - fn (str) – filename of the ENERGIES file (can include path to the file).
- code –
List of codes written as strings describing which information should be extracted from the file. The available codes are:
'steps' : Step indices 'E_kel' : Electronic kinetic energy (only for CPMD) 'Temp' : Temperature [K] 'E_KS' : Kohn-Sham energy [a.u.] 'E_cla' : Classical energy, E_KS + E_kin (constant for BOMD) 'E_ham' : 0 for BOMD 'RMS' : Nuclear displacement wrt initial position (?) 'CPU_t' : CPU time
- factor (int) – Integer factor used to skip some information. E.g. for an MTS calculation the high level information can be extracted by setting factor equals to the MTS factor used in the calculation. Default value is 1 (every time step info is extracted).
- HIGH (bool) – define wether to extract the information every
factorstep (this is default, HIGH=True) or the negative counter part meaning the info is extracted for every step except every factor steps HIGH=False.
Returns: The function returns a dictionary with keys the input codes and with values an array (
np.array) containing the corresonding information asfloats. The exception is the value for'steps'which is a simple list of integers.For example if the input codes are
code=['steps','E_cla'], the output dictionary will have the form:>>> read_ENERGIES('ENERGIES', ['steps','E_cla']) {'E_cla': array([-546.99550862, -546.99770079, ..., -546.96549996]), 'steps': [1, 2, ..., 10000]}
-
read_SH_ENERG(fn, nstates=None, factor=1)[source]¶ Read SH_ENERG.dat file and exctract info in dictionary
-
read_MTS_EXC_ENERG(fn, nstates, MTS_FACTOR, HIGH)[source]¶ Read MTS_EXC_ENERG.dat file and exctract info in dictionary.
Either the HIGH or the LOW level info will be exctracted.
-
xyz_to_cpmd_atoms(xyz_data=None, xyz_filename=None, PPs=None)[source]¶ Return list of strings as in CPMD ATOMS section.
qmmm_utils¶
Set of routine to deal with xyz data of QMMM calculations.
In particular for solute/solvent system it is possible to extract a QM region based on a distance criterion from the solvent.
-
read_xyz_structure(xyz, nat_solute, nat_solvent)[source]¶ decompose xyz data as solute + group of solvent molecules
-
get_QM_region(xyz, nat_slu, nat_sol, nmol_sol)[source]¶ Extract QM information from the full set of coordinates.
The following structure is assumed:
First
nat_sluatoms are the atoms of the solute molecules The rest are the solvent molecules arranged in blocks. For example, for a water solvent (nat_sol = 3):O ... H ... H ... O ... H ... H ... :
Parameters: - xyz (list) – xyz_data (list of list) for the whole system. Each sublist contains an atomic symbol and 3 (x,y,z) coordinates.
- nat_slu (int) – number of atoms of the solute molecule.
- nat_sol (int) – number of atoms of a single solvent molecule.
- nmol_sol (int) – total number of solvent molecules that should be included in the QM region.
Returns: xyz_QM, atom_types, max_dist
xyz_QM (list): xyz_data for the QM region.
- atom_types (dic): Each key is an atomic symbol and the
corresponding value is a list of indices of the atom of that type present in the QM region. The indices refer to the ordering of the original xyz_data.
- max_dist (float): Distance (in AA) between the center of
mass of the solute and the center of mass of the farthest solvent molecule included in the QM region
mts_md_turbo¶
Multiple time step algorithm for Molecular Dynamics with Turbomole.
Default usage¶
- The following files are required:
- GEOMETRY: a CPMD file for starting positions and velocities.
- define_high.inp: Input to TURBOMOLE’s define script to set up
- the high level electronic structure calculations.
- define_low.inp: same as define_high.inp but for low level.
From a python script or interpreter:
# setup the system, the list of atoms needs to be the same as in the GEOMETRY file.
system = system_settings(['O','H','H'])
# Run MD
data = md_driver(system)
The output dictionary data, contains the positions, velocities, forces, and energies
along the MD trajectory.
The MD and TURBOMOLE parameters can be modified by setting the md_settings and
turbo_settings objects and passing them to the md_driver.
-
md_driver(sys_in, md_in=<comp_chem_utils.mts_md_turbo.md_settings object>, turbo_in=<comp_chem_utils.mts_md_turbo.turbo_settings object>)[source]¶ Run a molecular dynamic using the RESPA algorithm of Tuckerman.
-
initialization(fname='GEOMETRY', nmts=1)[source]¶ Initialize positions, velocities, forces, and energies.
The initial positions and velocities are read from a CPMD GEOMETRY file in atomic units. (Bohr not Angstrom!)
The Low and High level forces and energies are then calculated by calling the Turbomole program.
-
get_forces_and_energy(x, level)[source]¶ Calculate forces and energies at a given level by calling TURBOMOLE.
-
data_update(data, x, v, f_low, f_high, e_low, e_high, idx, nmts)[source]¶ Update the data dictionnary with the current iteration values.
The data are also written to disk.
periodic¶
Simplified version of periodic table of atomic elements
molecule_data¶
Set of classes to deal with molecular data and formats
amino_acids¶
Just a dictionary of amino acids
pdbfile¶
Suppose to deal with pdb file format
-
class
pdb_file[source]¶ for now we assume a very simple structure: CRYST1 ATOM ATOM … END ATOM ATOM …
-
change_atom(new_name, new_symb, old_name, old_symb)[source]¶ change the atom name and symbol for a specific type of atom in the pdbfile
-
-
class
structure[source]¶ for now we assume a structure is just a list of residues
-
change_atom(new_name, new_symb, old_name, old_symb)[source]¶ change the atom name and symbol for a specific type of atom in the structure
-
-
class
residue[source]¶ -
-
change_atom(new_name, new_symb, old_name, old_symb)[source]¶ change the atom name and symbol for a specific atom in the residue
-
mysql_tables¶
Definitions of classes to work with molecular data.
The mol_info_table class is used to store and manipulated information about a molecule.
The mol_xyz_table class is used to store and manipulated the xyz coodinate of a molecule.
The functions defined here are basically wrappers to MySQL execution lines. Each function returns a MySQL command that should be executed by the calling routine.
program_info¶
read_gaussian¶
This file contains a set of functions usefull to extract some information from Gaussian output files.
-
get_gaussian_info(filename)[source]¶ Extract relevant information from a Gaussian output file.
Goes through a gaussian output file looking for the information required to calculate PDOS.
Parameters: filename (str) – Name of the Gaussian output file, it may also include the path to the file. Returns: Number of occupied orbitals. nbas (int): Number of basis functions (atomic orbitals).
overlap (np.array): Squared AO overlap matrix.
epsilon (list): Molecular orbital energies.
coef (np.array): Matrix of the molecular orbital coefficients.
Return type: nocc (int)
References¶
| [Barbatti2010a] | Barbatti, M. et al., (2010). The UV absorption of nucleobases: semi-classical ab initio spectra simulations. Physical Chemistry Chemical Physics, 12, 4959. http://doi.org/10.1039/b924956g |